![]()
DIA-Umpire is an open source Java program for computational analysis of data independent acquisition (DIA) mass spectrometry-based proteomics data. It enables untargeted peptide and protein identification and quantitation using DIA data, and also incorporates targeted extraction to reduce the number of cases of missing quantitation.
Four Major Modules
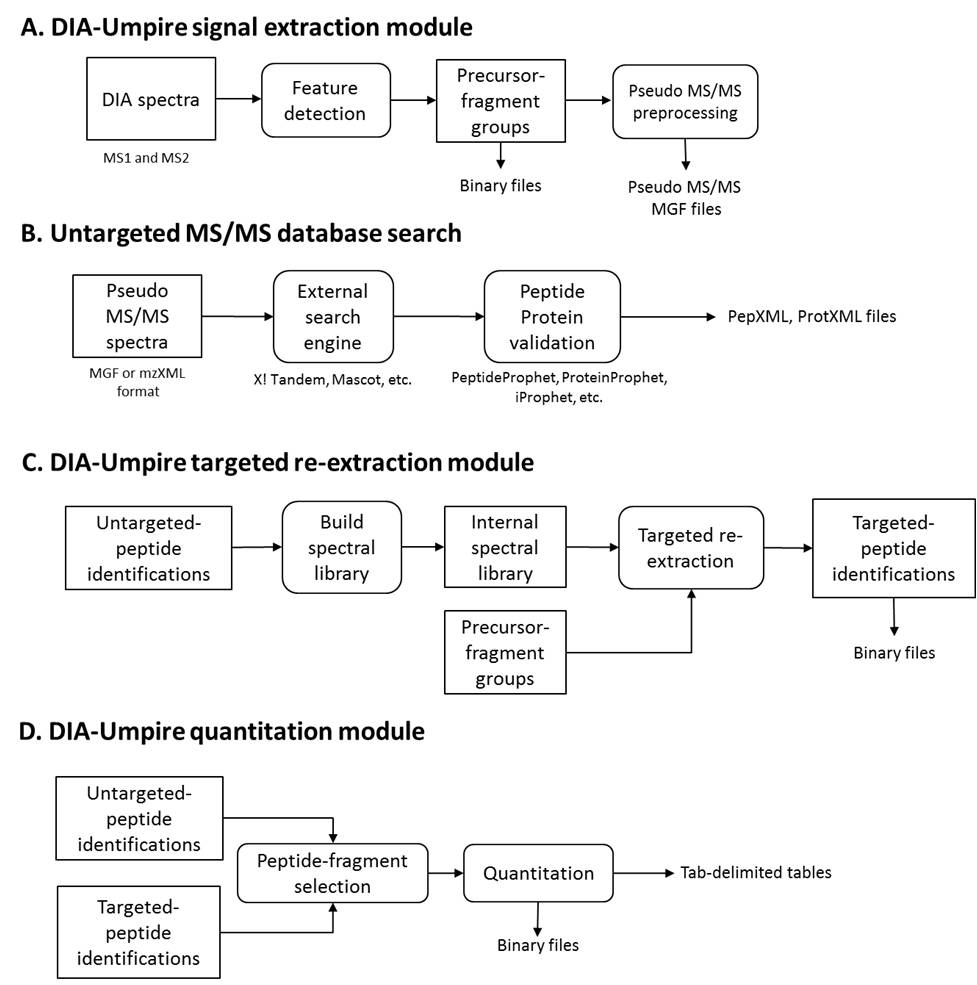
Suggested workflow
Depending on the scale of applications, three application scenarios which require different combinations of DIA-Umpire modules are described in our user guides.
-
Identification only analysis (Steps A->B)
-
Small scale identification and quantitation analysis with minimal computational costs (Steps A->B->D)
-
Complete DIA-Umpire identification and quantitation analysis (Steps A->B->C->D)
System Requirement
DIA-Umpire is written in Java, which is cross operating system programming language. To execute DIA-Umpire, Java 7 or higher (download link:Java SE Runtime Environment 7) version is required. As a rule of thumb, it is recommended have at least double the amount of RAM as the average size of your mzXML files (mzXML written in 32-bit format without zlib compression). If mzXML is in 64-bit format, then RAM requirements should be approximately the size of the file.
Other Usages
DIA_Umpire_SE
The first step of DIA-Umpire analysis. It is a signal extraction module to generate pseudo MS/MS spectra given a DIA file
- java -jar -Xmx8G DIA_Umpire_SE.jar mzMXL_file diaumpire.se_params
DIA-Umpire
Main DIA-Umpire class libraries
- ExternalPackages: external packages, currently including JAligner, SortedListLib, JMEF, and a traML parser developed by ISB.
- FDREstimator: wrapper to generate FDR filtered protein and peptide lists
- MSUmpire: Umpire libraries
- BaseDataStructure : basic data structure, including x-y pair value data, scan data, and parameter setting class etc.
- DIA : DIA specific classes
- FragmentLib : Fragment library manager, basically it’s a spectral library
- LCMSPeakStructure : Data structure classes for MS1 or MS2 peak for to a LCMS run
- MathPackage : Math calculation classes
- PSMDataStructure : Data structure classes related to Mass-spec based identifications, ranging from PSM, peptide ion, protein and identification data structure for a LC-MS run (LCMSID.java). In addition, this package includes processing manager to extract PTM and peptide fragment information generated from Compomics library.
- PeakDataStructure : Data structure classes related to peak data, from peak curve, peak isotope cluster, and peak smoothing algorithms.
- PeptidePeakClusterDetection : Processing classes to detect peak features
- SearchResultParser : PepXML, ProtXML parsers
- SeqUtility : Classes for sequence processing, including FastaParser and shuffled sequence generator
- SpectralProcessingModule : Spectrum peak processing classes
- SpectrumParser: mzXML, mzML parsers
- Utility: Other classes
- resource: all resource files in text format
MS1Quant
DDA-based MS1 quantification tool based on the feature detection algorithm.